ff_map¶
The ff_map subprogram takes a trajectory at a higher resolution (such as atomistic),
and converts it to a trajectory at a lower resolution (such as coarse-grained Martini).
The resultant trajectory is known as a pseudo-atomistic one, as it represents the position of
the beads as if they exactly replicated an atomistic conformation.
System preparation¶
We start with an atomistic trajectory of the peptide-like molecule, GSH, performed using the Charmm36
force field. After running a full production trajectory, the trajectory has been pbc corrected and centered.
Mapping solvent molecules is not currently possible for ff_map, so this is an
essential step in preparing your system. Such a processing could be performed using the following Gromacs commands:
# process the trajectory
gmx trjconv -f prod.xtc -s prod.tpr -pbc mol -center -o atomistic.gro -e 0
gmx trjconv -f prod.xtc -s prod.tpr -pbc mol -center -o atomistic.xtc
The files in the AA
subfolder called atomistic.gro, atomistic.xtc and atomsitic.tpr are readily prepared solvent-free atomistic coordinate,
trajectory and topology files for use for the rest of this tutorial.
Preparing the map file¶
The solvent free map file can now be used to prepare a map file using the cgbuilder tool. The coordinate can be uploaded, and beads assigned to atoms by clicking on the interactive visualisation pane. Beads should be assigned following the standard Martini mapping rules (covered for example, here.)
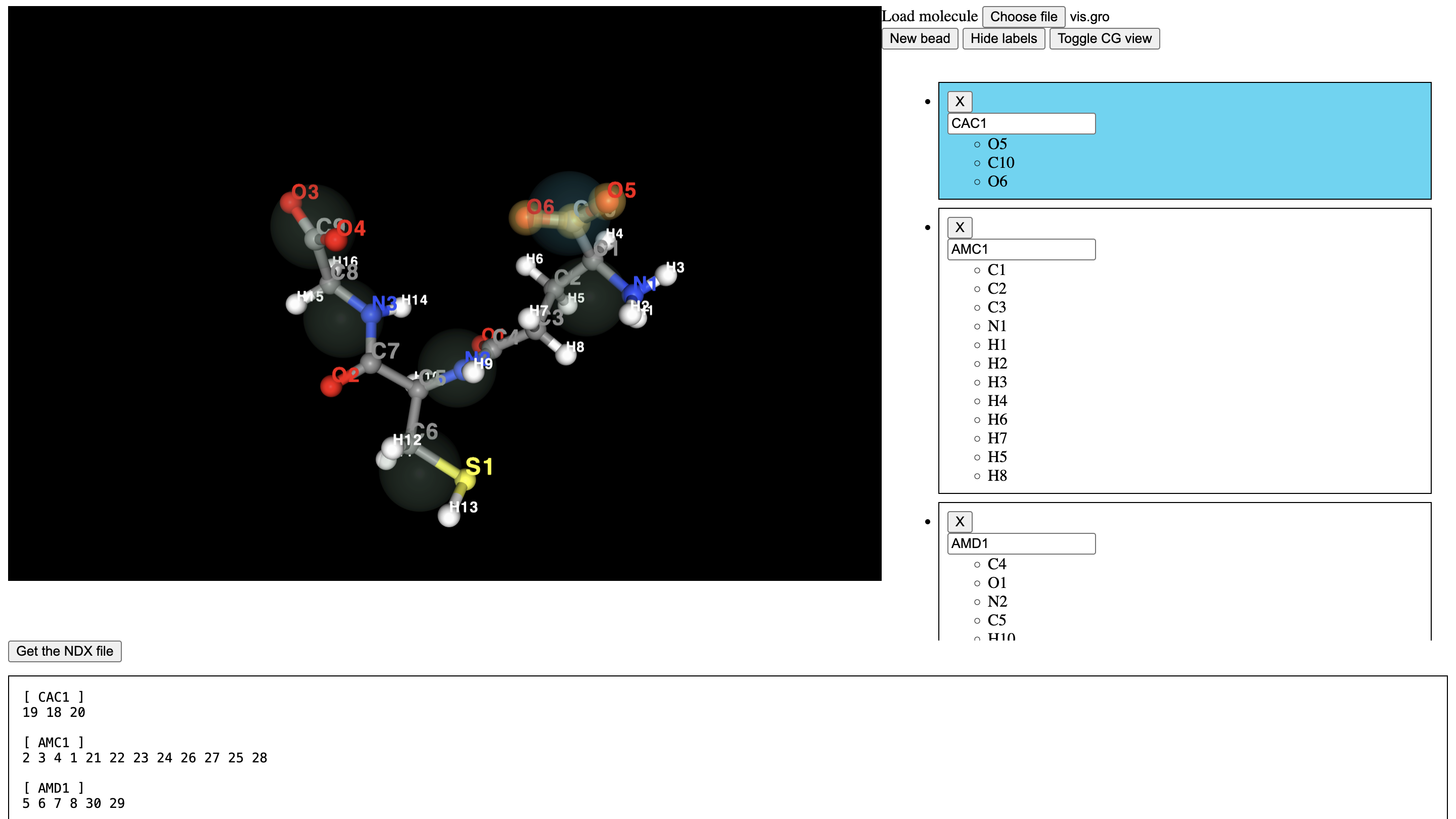
The .map file can be downloaded further down the page, or the example file in the data directory can be used.
A small modification required is the header directive, which must be changed to the following:
[ molecule ]
LIG GSH
Here, the molecule directive indicate residue names in the topology file at atomistic resolution (LIG)
and the name at CG resolution (GSH).
Mapping the molecule¶
With the .map file prepared, we can map the system. The trajectory can be mapped simply with:
ff_map -f atomistic.xtc -s atomistic.tpr -m GSH.map -o mapped.xtc -mols LIG
Note that by mapping a trajectory, the first frame is written as a simple coordinate file (.gro) also
with the same file name prefix (in this case, it will be called mapped.gro).
Use of the -mols flag
The -mols flag here is the moleculename of the molecule as described in the topology. In this case,
the fact that the molecule consists of only one residue, also called LIG, is a coincidence.
The mapped structure could now be viewed in vmd to check that everything is now in place:
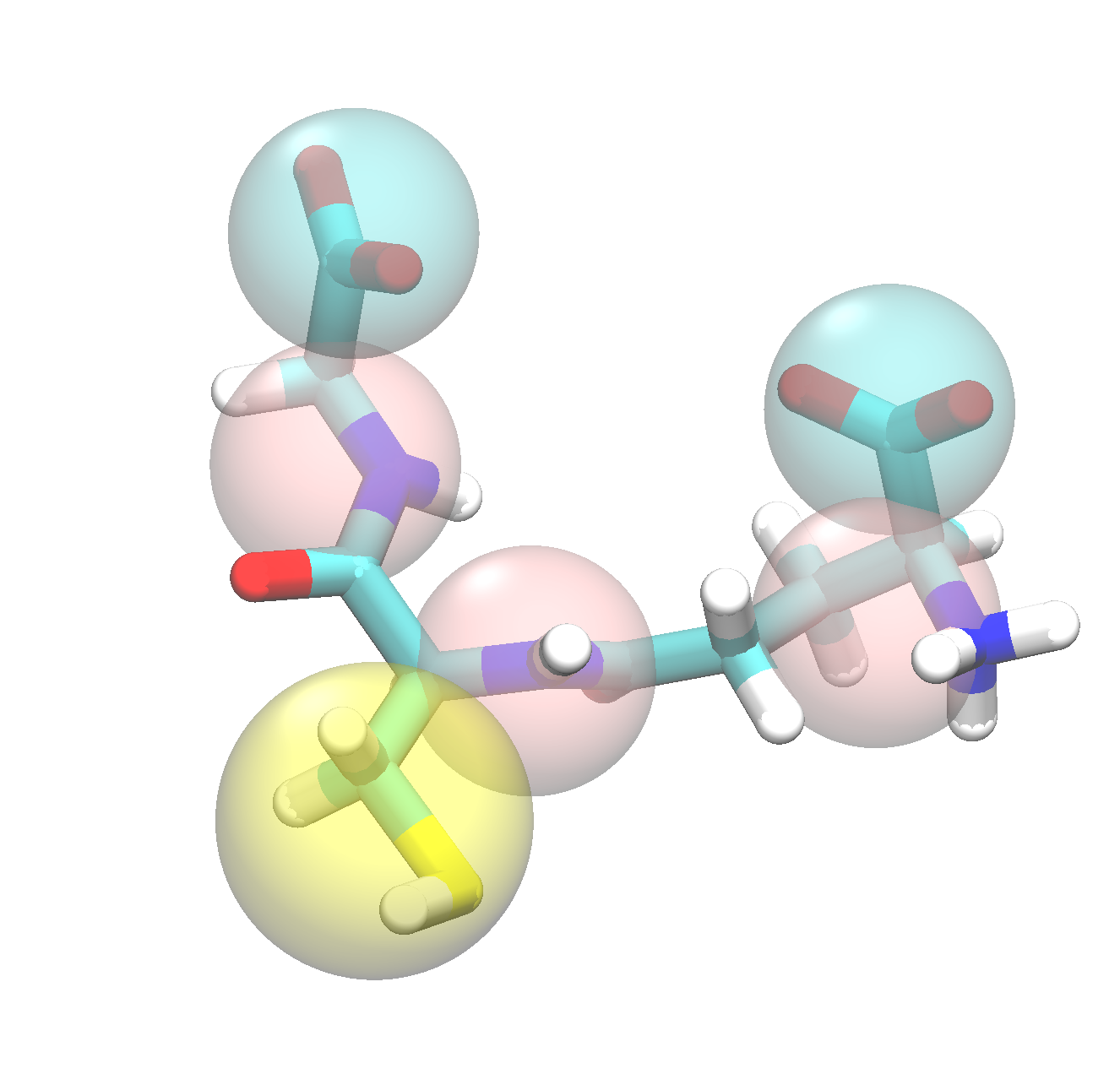
The tutorial folder data/CG contains the mapped first frame and trajectory.
From here, the mapped trajectory can be used in the ff_inter subprogram to generate
initial parameters for the molecule.