ff_inter¶
Background¶
To parameterise a molecule for the Martini force field, effective interactions between
beads need to be converted from the underlying mapped interactions between groups of atoms.
This can be achieved in fast forward using the ff_inter program, which takes a
mapped trajectory together with a topology file describing the system to calculate
the distributions of the interactions.
Options¶
ff_inter has the following options:
-f TRAJFILE trajectory file (default: None)
-s TPRFILE tpr file (default: None)
-n NDX_FILE index file (default: None)
-pref PREFIX common prefix to filename (default: )
-i [ITP_FILES ...] itp file (default: None)
-dists [DISTRIBUTION_DATA]
Save text files with time series and distribution data for interactions. Optionally provide a
path (default: current dir). (default: None)
-plots [PLOTS] Plot the reference and fits of bonded interactions. Optionally provide a path (default: current
dir). (default: None)
-plot-data Save a pickle file containing the input data for the plots (default: False)
-constraints CONSTRAINT_CONVERT
Force constant above which to convert bonds to constraints (default: 10000)
-temperature TEMPERATURE
Temperature to use in Boltzmann inversion (default: 310)
-precision PRECISION Precision of variables written to itp (default: 3)
-max-dihedral MAX_DIHEDRAL
Maximum multiplicity of dihedral to try stacking during dihedral fitting (default: 10)
-dihedral-scaling DIHEDRAL_SCALING
Scale factor for strength of proper dihedral terms from fit to potential (default: 1000.0)
-interactions {guess,comments,all}
Interactions (angles and dihedrals) to fit. Will use only commented interactions in itp by default, or all interactions if 'all' is specified. Will guess interactions from the bonds if 'guess'
is specified. (default: comments)
-dist-matrix Save text files with time series and distribution data for all pairwise bead distances
(default: False)
-lincs Estimate the LINCS order required for solving the constraints in the molecule(s) (default:
False)
Example¶
ff_inter -f mapped.xtc -s mapped.tpr -i molecule.itp -interactions comments -dists -dist-matrix -plots -lincs
In the above command, the ff_inter subprogram takes the mapped/pseudo-atomistic system described by mapped.tpr
and mapped.xtc, and interrogates the bonded interactions of the molecule described by molecule.itp. On
completion, the subprogram will write out a new itp file (backing up the input one), with bonded parameters
derived from fittings to the interaction distributions in the input trajectory.
Note
What is an interaction and what do we mean by interrogating it?
Consider two coarse grained beads in the mapped trajectory that have a chemical bond between them in the coarse grained topology. A fundamental question for parameterising the molecule is How far away are these two beads from each other, and so how long is the bond between them? How stiff should the resultant bond be? One can extend this for further groups of three or four atoms with bonds between them, and ask the same question for angle and dihedral potentials in a coarse grained topology.
By indicating that we want to investigate these interactions in the input topology file, we tell the ff_inter
subprogram to firstly generate time series data of these sets of beads, and then fit to time-averaged distributions,
By using the comments mode of the -interactions flag, only the interactions which have been specified by the
comments in the given itp file will be investigated and modified. More details and further options for this flag are
given below.
The -dists and -dist-matrix flags ensure that .dat files are written out for each interaction and
inter-bead distance in the molecule. These files are designed to be compatible later with the ff_assess subprogram.
Both the time series data file, and a time-averaged distribution of the data are written.
The -plots flag generates a plot of the time-averaged interaction distributions, also showing how the subprogram has
fitted them. This may be useful for debugging poorly-fitted parameters during later optimisation.
Of these options, the trajectory, topology, and itp files are strictly necessary. Other options are described below.
itp file format¶
Denoting interactions¶
Interactions can be computed from one or multiple itp files. The molecule name
is simply matched to those found in the tpr and trajectory file and then the
interactions are computed. Using the -interactions flag, users can specify which interactions to compute.
By default or when using the -interactions comments flag, only interactions with a comment at the end of the line are computed.
This allows users to specify which interactions to compute and to group together repeated interactions by using the same comment.
For example, from the following entry the first two bonds are put in a group and computed together and the last bond is skipped.
[ bonds ]
1 2 1 2000 ; group1
2 3 1 2000 ; group1
4 5 1 1500 ; group2
4 5 1 1000
With the -interactions all flag, all interactions in the input itp file are computed and modified in the output itp file,
regardless of whether they have comments or not.
With the -interactions guess flag, interactions are guessed from the bonds in the input itp file.
This may be useful for parameterising a single molecule with a minimal itp file describing only atoms and bonds, as described below.
Minimal itp files¶
ff_inter can generate all interactions implicitly from minimal itp files describing only
atoms and bonds, with the -interactions guess flag. This may be useful for parameterising a single molecule.
In this case, the input itp file can look like:
[ moleculetype ]
MOL 1
[ atoms ]
1 TYPE 1 RESNAME ATOMNAMEa 1
2 TYPE 1 RESNAME ATOMNAMEb 2
3 TYPE 1 RESNAME ATOMNAMEc 3
4 TYPE 1 RESNAME ATOMNAMEd 4
[ bonds ]
1 2 1 0.1 10000 ; ATOMNAMEa_ATOMNAMEb
2 3 1 0.1 10000 ; ATOMNAMEb_ATOMNAMEc
2 4 1 0.1 10000 ; ATOMNAMEb_ATOMNAMEd
3 4 1 0.1 10000 ; ATOMNAMEc_ATOMNAMEd
From a mapped trajectory for this molecule, ff_inter will write a new itp file for the
molecule containing fitted bonded parameters not only for bonds, but also for angles
and dihedrals that it automatically identifies resulting from the molecular graph.
File writing¶
ff_inter preserves existing itp files with identical names through safe file backups.
Distributions and plotting¶
Saving files¶
ff_inter computes time series and resulting distributions for input molecular topologies
for the interactions specified in itps. To save the text files, figures of fitted
distributions, and the data used for plotting, the -dists, -dist-matrix, -plots and -plot-data
arguments can be used respectively. If the -prefix argument is given, it is used for
all file names in the output.
Two kinds of distribution files are written, designed in part for subsequent use with ff_assess.
Plots¶
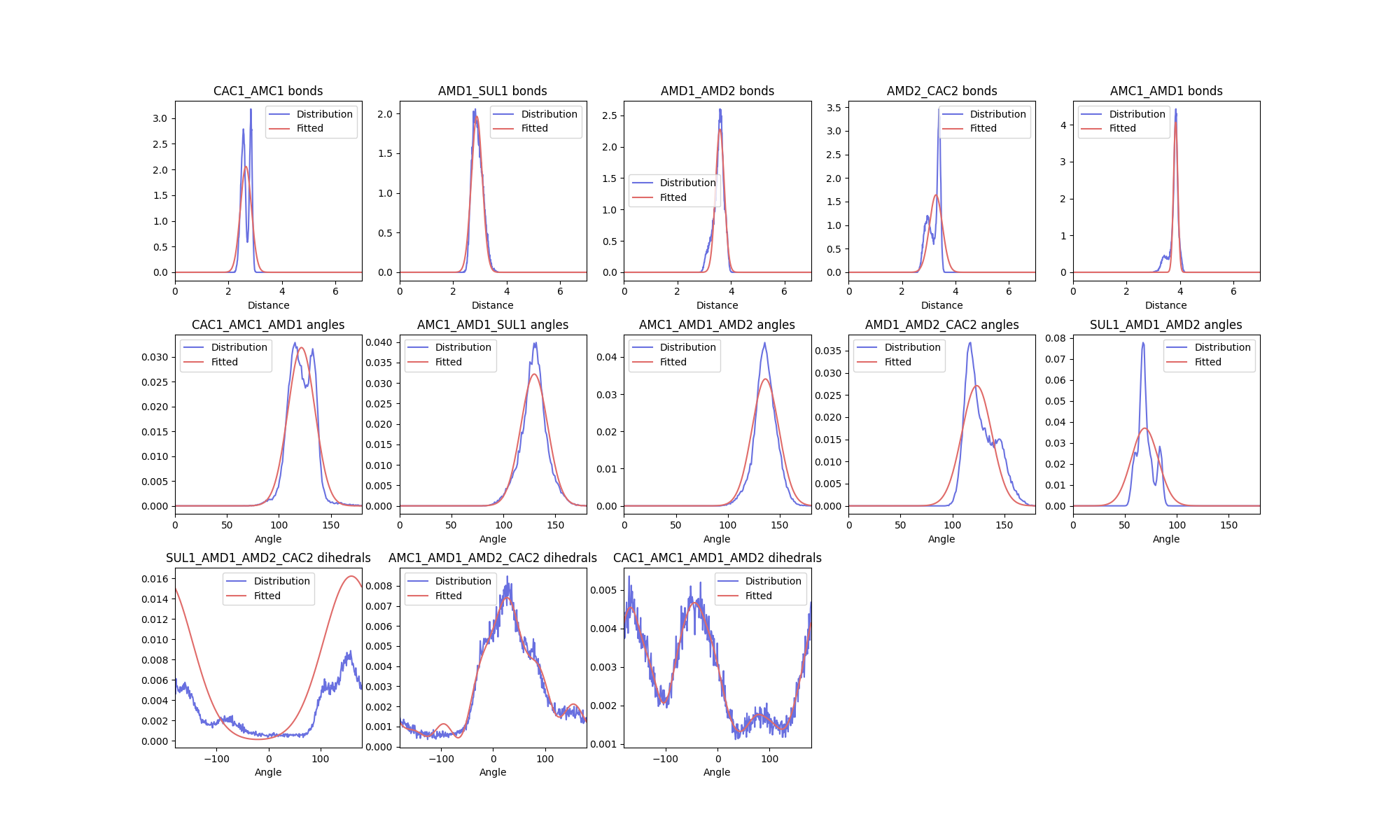
The figure shown is an example of a distribution plot generated by ff_inter. Each subplot
has a title corresponding to interaction type and the atoms involved. For each interaction
in the plot, the raw data (in blue) is plotted along with the fitted distribution (in red).
Plot data¶
The plot data is saved as a pickle file. The pickle file contains a nested dictionary of
all the data required to generate the output plot, for users’ own analysis and use.
The dictionary is structured to group individual interactions by their interaction type.
Each entry contains items to reproduce the plot, as well as basic fitting information. For
example, the angle entry shown below contains the arrays of the mapped distribution (Angle
and simulated_distribution), as well as the fitted distribution determined by ff_inter
(fitted_distribution). Finally it contains entries for the center and sigma
(distribution_center and distribution_sigma) of the fitted distribution used to
determine the parameters in the output itp file.
{
"angles":{
"ATOMNAMEa_ATOMNAMEb_ATOMNAMEc":
{
"Angle": [0.5, 1.5, 2.5, ... ],
"distribution_center": 80.173,
"distribution_sigma": 12.99018238723656,
"fitted_distribution": [2.0831593372894023e-10, 3.330336459711729e-10, 5.292733885907642e-10, ... ],
"simulated_distribution": [0.0, 0.0, 0.0, ...]
},
"ATOMNAMEb_ATOMNAMEc_ATOMNAMEd":
{
"Angle": [0.5, 1.5, 2.5, ... ],
...
}
...
}
"bonds":{...}
...
}
Dihedral fitting¶
One of the powerful features of ff_inter is the fitting of multiple functions to mapped
dihedral distributions. ff_inter will fit dihedrals of various multiplicities to fit
the underlying distributions to fit the form of proper dihedral used by gromacs.
ff_inter will further try to discriminate between dihedrals of proper and improper types.
Based on fitting statistics, it will use either a series of increasing multiplicity
dihedrals or a gaussian function to fit the underlying data and determine the correct
output parameters for an itp file.
caveat emptor¶
While fitting multiple dihedrals may result in a faithful theoretical reproduction of
mapped dihedral distributions, it may not result in a production-stable molecule. The
-max-dihedral and -dihedral-scaling arguments may be useful to optimise a
molecule topology for these purposes.
By default, ff_inter will attempt to fit up to 10 dihedrals of increasing multiplicities
in order to best match the underlying mapped dihedral distribution:
With the -max-dihedral argument, the maximum number of contributing functions can be limited
to a value as indicated in the sum. While the fitted distribution may not result in as
faithful a match, it may improve the stability of a molecule for simulation purposes.
The -dihedral-scaling argument may be similarly useful in preparing stable molecules. The
values of \(k_n\) in the above description of the final function may be sufficiently
weak as to have little-to-no effect on the dihedral distribution during simulation. Therefore,
these factors are scaled by a fixed value when written to itp files. Nominally, this is 1000,
but this can be changed using the -dihedral-scaling argument.
Constraints¶
If the force constant of a bond is very high, it is usually better to convert it to
a constraint.
ff_inter sets this value to be 10,000 kJ/mol/nm^2 by default, but this can be changed using
the -constraints argument. The -lincs argument can also be used to estimate the LINCS order for solving the
constraints during the simulation.